 手機版
手機版 化工儀器網手機版
化工儀器網手機版
 化工儀器網小程序
化工儀器網小程序
 官方微信
官方微信 公眾號:chem17
公眾號:chem17
 掃碼關注視頻號
掃碼關注視頻號











【應用】Sepmatix 8x平行色譜系統助力藥物合成產物篩選色譜柱

Sepmatix 8x平行色譜系統
助力藥物合成產物篩選色譜柱
色譜應用
”
1
簡介
藥品屬于一類化學或生物來源的產品,用于人類或動物的醫療。化學合成常用于藥物的生產或潛在藥物的開發。化學合成是一個經常與雜質存在有關的過程,因為收率很少是100%。這些雜質會對最終產品的功效、安全性和質量產生重大影響。因此對藥物進行純化以保證合成化合物的純度和完整性至關重要,藥物的純化通常可以用色譜法進行。
近年來,超臨界流體色譜(supercritical fluid chromatography,SFC)作為一種替代反相液相色譜(RP-LC)的方法出現了。SFC結合了氣相色譜和液相色譜的優點。SFC使用超臨界二氧化碳作為流動相的一部分,這是一種清潔和綠色的溶劑,很容易從最終產品中去除。此外,SFC提供了高分辨率和快速分離。
與RP-LC相比,C18柱對大多數樣品至少達到可接受的選擇性水平,在SFC中沒有通用的通用固定相。在SFC中需要對不同的固定相進行篩選,以確定待分離樣品的最佳選擇性。二氧化碳的低極性允許非極性以及更多極性固定相整合到篩選過程中。確定最佳固定相并連續實施制備方法后,可對樣品混合物進行純化。
在本文中,合成了不同的藥物并純化它們。第一步,篩選不同的固定相,確定每種情況下的理想選擇性。在第二步中,描述了用于實施制備純化方法的過程。
2
實驗設備
-
Sepiatec SFC-50
-
Sepmatix 8x SFC
-
PrepPure Silica, 5 um, 250 x 4.6 mm
-
PrepPure Diol, 5 um, 250 x 4.6 mm
-
PrepPure Amino, 5 um, 250 x 4.6 mm
-
PrepPure 2-EP, 5 um, 250 x 4.6 mm
-
HILIC, 5 um, 250 x 4.6 mm
-
PrepPure PEI, 5 um, 250 x 4.6 mm
-
PrepPure CBD, 5 um, 250 x 4.6 mm
-
Cyano, 5 um, 250 x 4.6 mm, (Dr. Maisch GmbH)
-
PrepPure 2-EP, 5 um, 250 x 10 mm
-
PrepPure PEI, 5 um, 250 x 10 mm
-
PrepPure Amino, 5 um, 250 x 10 mm
-
PrepPure Diol, 5 um, 250 x 10 mm
3
試劑和材料
-
普魯卡因合成產物
-
二氧化碳 (99.9%)
-
甲醇 (≥ 99%)
-
2M氨氣溶于甲醇中
-
甲酸 (99%)
-
去離子水
-
對乙酰氨基酚合成產物
-
利多卡因合成產物
-
乙酰水楊酸合成產物
-
苯佐卡因合成產物
為了安全操作,請注意所有相應的MSDS!
4
實驗過程
運行條件 Sepmatix 8x SFC:
-
流動相:A =二氧化碳;B=甲醇
-
色譜柱尺寸:250 x 4.6 mm
-
流速:3ml /min(每根色譜柱)
-
流動相條件:0-0.5 min: 5% B
-
0.5-8.0 min:5-50% B
-
8.0-9.4 min:50% B
-
9.4-9.5 min:50-5% B
-
9.5-10 min:5% B
-
檢測:200nm - 600nm 紫外掃描
篩選運行是完全自動開始的。使用流量控制單元將流量設置為每通道3ml /min,并平衡色譜柱。自動進樣(V = 5 μL),開始平行篩選(運行時間= 10 min)。背壓調節器設置為150 bar,柱箱加熱至32°C。添加劑需要提前被添加到改性劑中。
SFC-50 運行條件:
-
流動相:A =二氧化碳;B=甲醇
-
柱尺寸:250 × 10mm
-
流動相條件:等度運行
-
檢測:紫外
SFC柱在規定的流速下條件平衡3分鐘。使用定量環自動進樣,并開始運行。背壓調節器設置為150 bar,柱箱加熱至40°C。添加劑需要提前被添加到改性劑中。
5
實驗結果
5.1 對乙酰氨基酚
對乙酰氨基酚(Acetaminophen,AA),也被稱為撲熱息痛,是一種鎮痛、解熱和非手性藥物。它屬于非阿片類鎮痛藥。化學上可以通過對氨基酚(AP)與乙酸酐反應合成,其中N-乙酰化發生(見圖1)。為了確定對乙酰氨基酚合成產物純化的理想選擇性,第一步進行了色譜柱篩選(見圖1)。


▲ 圖1. 上:對乙酰氨基酚合成反應方程,下:篩選結果Sepmatix 8x SFC;從左到右色譜柱填料依次為:硅膠、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;運行時間= 10分鐘。
共洗脫發生在二醇相和 2-EP 相。硅膠,CBD,氰基和氨基相沒有表現出理想的選擇性,因為沒有基線分離可以在這里實現。HILIC和PEI相表現出良好的選擇性和分辨率,因為分辨率總是高于1.5(見表1)。1.5的分辨率意味著兩個峰可以很好地分離。表1 也顯示了洗脫順序。氰基相呈現出相反的洗脫趨勢,對氨基酚先被洗脫,對乙酰氨基酚后被洗脫。篩選結果表明,該反應并非 100% 完成,產物中仍存在大量對氨基酚。
表1. SFC 條件下不同篩選柱的分辨率值和洗脫順序
色譜柱 | R | 洗脫順序 |
硅膠 | 0.95 | AA,AP |
氨基 | 0.58 | AA,AP |
氰基 | 1.62 | AP,AA |
二醇基 | X | X |
2-EP | X | X |
HILIC | 3.04 | AA,AP |
PEI | 5.57 | AA,AP |
CBD | 1.05 | AA,AP |
選擇 PEI 色譜柱進行制備純化,因為它具有最高的分辨率(見圖2)。根據篩選得到的色譜圖,利用梯度法可以確定 AA 和 AP 洗脫時流動相中甲醇的近似含量。該改性劑含量可作為等度制備方法的指導值。AA 和 AP 洗脫液中改性劑含量在 35-40% 之間。圖2(上)為改性劑含量為 40% 時,在制備 PEI 柱上對AA的純化。AA 和 AP 可以很好地分離。在相同條件下,可采用疊層進樣法對 AA 進行自動化純化(見下圖2)。因為只有AA是相關的,所以不收集 AP。


▲ 圖2. AA純化的單次進樣(上)和疊層進樣(下);運行條件:流量= 30 mL/min,改性劑為甲醇,改性劑% = 40%,溫度= 40℃,背壓調節器=150 bar,進樣量= 250 μL,紫外波長= 254 nm;疊層進樣條件:注射次數= 10次,疊層時間= 1.8 min,色譜峰收集 = 1(以時間為基礎)。
5.2 利多卡因
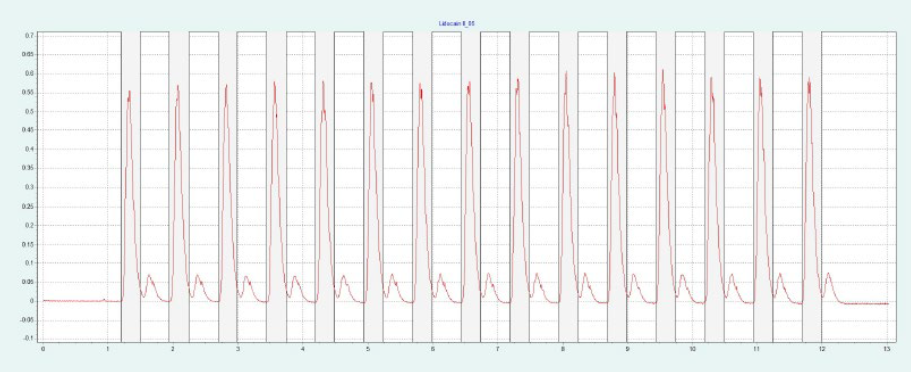
利多卡因(L),化學式為2-二乙胺- N -(2,6-二甲基苯基)乙酰胺,是一種用作局部麻醉劑和抗心律失常藥物的藥物。它起到鈉通道阻滯劑的作用。利多卡因可以兩步合成(見圖3)。在第一步中,2,6 二甲醚(X)的氨基被酰化。在第二步中,中間產物(IP)通過與二甲胺的親核取代轉化為利多卡因。因此,需要兩個純化步驟。色譜柱篩選的結果如 圖2 所示。在改性劑甲醇中始終加入 20mm 氨水作為堿性添加劑。


▲ 圖3. 上:利多卡因合成反應方程,下:篩選結果Sepmatix 8x SFC;從左到右依次為:硅膠、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;運行時間= 10分鐘
原則上,任何篩選的相都可以用于 IP 的純化,因為總是可以實現基線分離。氨基相在改性劑含量相對較低的情況下具有最高的分辨率。X 和 IP 的洗脫順序在每一相中都是恒定的。對于L的純化,氰基和 CBD 相沒有達到基線分離(見表2)。同樣,氨基相顯示出最高的分辨率。硅膠相是唯一表現出反洗脫趨勢的相。否則,L 總是先洗脫。篩選結果表明,不可能每一步都達到 100% 的產率。這是不尋常的合成 IP,因為酸性氯化物是高活性的。據推測,在合成過程中,化學計量學出現了錯誤,導致 X 的添加過量。
表2. SFC 條件下不同篩選柱的分辨率值和洗脫順序
色譜柱 | R1 | R2 | 洗脫順序1 | 洗脫順序2 |
硅膠 | 3.04 | 2.84 | X, IP | IP, L |
氨基 | 10.29 | 11.45 | X, IP | L, IP |
氰基 | 4.9 | 1.2 | X, IP | L, IP |
二醇基 | 3.35 | 3.44 | X, IP | L, IP |
2-EP | 4.18 | 5.67 | X, IP | L, IP |
HILIC | 7.63 | 6.33 | X, IP | L, IP |
PEI | 4.91 | 7.34 | X, IP | L, IP |
CBD | 2.81 | 1.12 | X, IP | L, IP |
選擇氨基色譜柱進行制備性純化,因為它具有最高的分辨率(見圖4)。篩選結果表明,X 和 IP 的洗脫量約為 10-19%,L 和 IP 的洗脫量約為 11-19%。圖4a 為改性劑 16% 時純化 IP 的制備性單次進樣,圖4b 為改性劑 20% 時純化L的制備性單次進樣。在相同的條件下,可以進行疊層進樣,分別對 IP 和 L 進行自動純化(見 圖4c 和 圖4d)。因為只有 IP 或 L 是相關的,所以不收集反應物。



▲ 圖4:單次進樣(a)和疊層進樣(c)純化IP;運行條件:流量= 20 mL/min,改性劑=甲醇+ 20 mM氨,改性劑% = 16%,溫度= 40℃,背壓調節器=150 bar,進樣量= 170 μL,紫外波長= 254 nm;疊層進樣條件:注射次數= 15次,疊層時間= 0.75 min,色譜峰收集= 1(以時間為基礎);L的純化采用單次進樣(b)和疊層進樣(d);運行條件:流量= 20 mL/min,改性劑=甲醇+ 20 mM氨,改性劑% = 20%,溫度= 40℃,背壓調節器=150 bar,進樣量= 170 μL,紫外波長= 254 nm;疊層進樣條件:注射次數= 20次,疊層時間= 0.65 min,色譜峰收集= 1(以時間為基礎)。
5.3 乙酰水楊酸
阿司匹林,化學成分為乙酰水楊酸(ASA),是一種眾所周知的鎮痛和抗炎藥。它屬于非甾體類抗風濕藥。它可以通過水楊酸(SA)與乙酸酐的化學反應合成,并發生酯化反應(見圖5)。為了確定乙酰水楊酸純化的理想選擇性,第一步進行了色譜柱篩選(見圖5)。
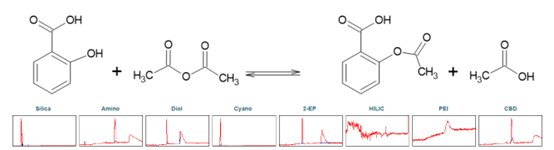
▲ 圖5. 上:乙酰水楊酸合成反應方程,下:Sepmatix 8x SFC篩選結果;從左到右依次為:硅膠、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;運行時間= 15分鐘。
由于 SA 在固定相中的高保留率,在梯度中添加 50% 改性劑 5 分鐘的等度分離以促進 SA 的洗脫。HILIC 相只有噪聲,ASA 和 SA 沒有洗脫。在 PEI 相中,實驗條件下只有 ASA 洗脫。除氰基相外,其余相均表現為 ASA 和 SA 的分離。2-EP 階段顯示了潛在運行時間和分辨率方面的最佳結果(參見表3)。篩選色譜圖顯示 ASA 合成沒有達到 100% 產率。
表3. SFC 條件下不同篩選柱的分辨率值和洗脫順序
色譜柱 | R | 洗脫順序 |
硅膠 | 1.68 | SA, ASA |
氨基 | 1.79 | ASA, SA |
氰基 | X | X |
二醇基 | 3.86 | ASA, SA |
2-EP | 6.85 | ASA, SA |
HILIC | X | X |
PEI | X | ASA |
CBD | 27.45 | ASA, SA |
選擇 2-EP 相進行制備純化。篩選結果表明,改性劑含量約為 26-40% 時,ASA 和 SA 可被洗脫。圖6(上)為改性劑含量為 40% 時,ASA 在 2-EP 制備柱上的純化結果。在改性劑中加入 0.5% 甲酸,改善 SA 的峰形。SA 在沒有甲酸的情況下顯示出很強的尾砂。隨著甲酸的加入,SA 的尾砂峰值降低。在相同條件下,可以采用疊層進樣法對 ASA 進行自動化純化(見下圖6)。因為只有 ASA 是相關的,所以不收集 SA 和其他雜質。
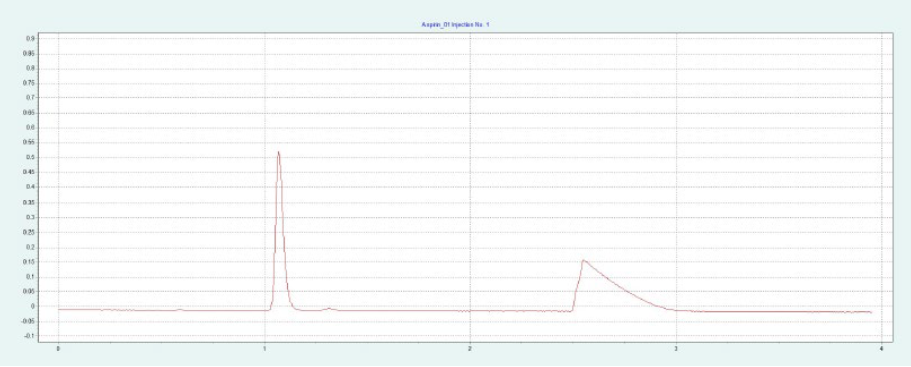
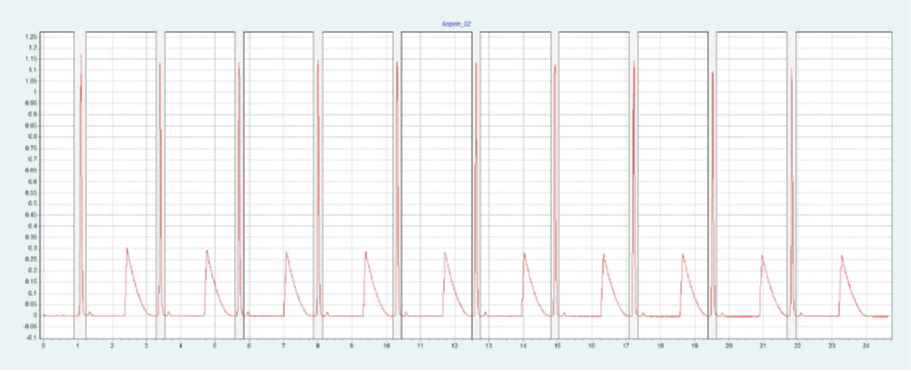
▲ 圖6. ASA純化的單次進樣(上)和疊層進樣(下);運行條件:流速= 30 mL/min,改性劑=甲醇+ 0.5%甲酸,改性劑% = 40%,溫度= 40℃,背壓調節閥 = 150 bar,進樣量= 200 μL,紫外波長= 254 nm;疊層進樣條件:注射次數= 10次,疊層時間= 2.0 min,色譜峰收集= 1(以時間為基礎)。
5.4 苯佐卡因
苯佐卡因(BC),化學名稱為 4-氨基苯甲酸乙酯,屬于局部麻醉劑。以對氨基苯甲酸(PABA)為原料,羧基與乙醇經酸催化酯化反應可合成對氨基苯甲酸(PABA)。硫酸被用作催化劑。進行柱篩選以確定苯佐卡因合成產物純化的理想選擇性(見圖7)。
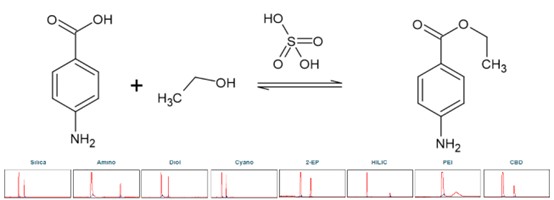
▲ 圖7. 上:苯佐卡因合成反應方程,下:篩選結果Sepmatix 8x SFC;從左到右依次為:硅膠、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;運行時間= 15分鐘。
由于PABA在固定相中的高保留率,在梯度中增加了一個在50%改性劑下5分鐘的等度步驟。由于酸性,用洗滌步驟除去了硫酸。各固定相的色譜圖表明,各固定相均能分離苯佐卡因。在洗脫速度和分辨率上存在較大差異(見表4)。苯佐卡因總是在PABA之前被洗脫。HILIC相和氨基相的分辨率最高,而PABA相的保留率很高,即使改性劑含量達到50%。色譜圖也表明酯化反應尚未完成。
表4. SFC 條件下不同篩選柱的分辨率值和洗脫順序
色譜柱 | R | 洗脫順序 |
硅膠 | 6.38 | BC, PABA |
氨基 | 21.6 | BC, PABA |
氰基 | 5.38 | BC, PABA |
二醇基 | 10.82 | BC, PABA |
2-EP | 10.86 | BC, PABA |
HILIC | 28.78 | BC, PABA |
PEI | 1.94 | BC, PABA |
CBD | 8.72 | BC, PABA |
選擇二醇相進行制備純化。HILIC 相和氨基相要求改性劑含量高,運行時間長。二醇相顯示樣品的快速洗脫,具有足夠高的分辨率。篩選結果表明,改性劑含量約為 30-40% 時,BC 和 PABA 均可洗脫。圖8(上)顯示了在改性劑含量為 28% 的制備二醇柱上純化 BC。在相同條件下,可以采用疊層進樣法對 BC 進行自動化純化(見下圖8)。因為只有 BC 是相關的,所以不收集 PABA。
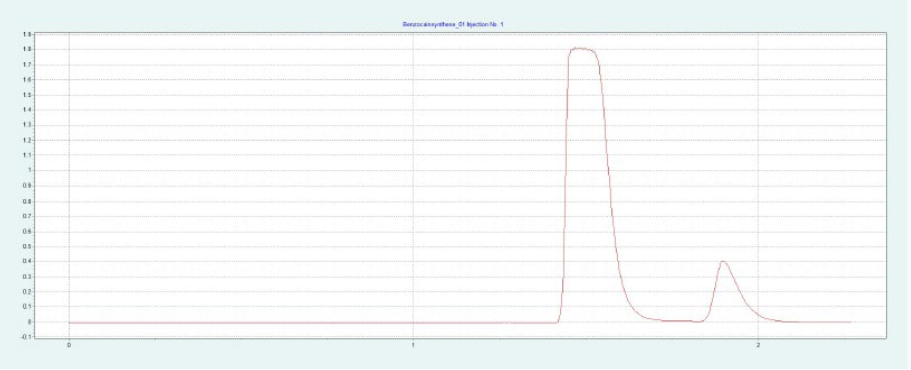
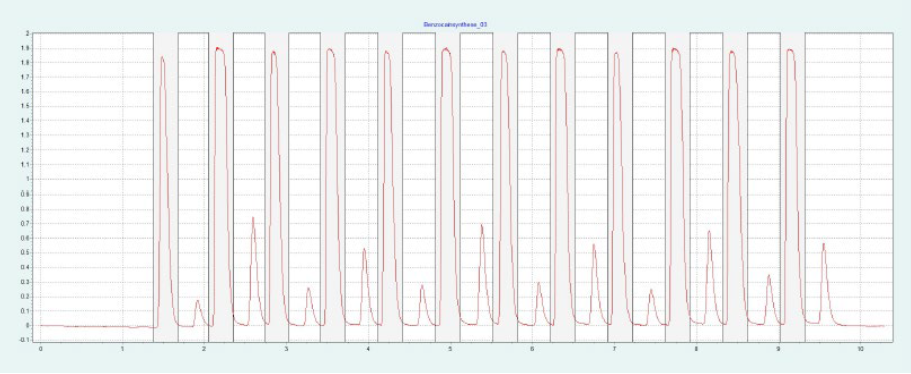
▲ 圖8. 苯佐卡因純化的單次進樣(上)和疊層進樣(下);運行條件:流速= 20 mL/min,改性劑為甲醇,改性劑% = 28%,溫度= 40℃,背壓調節器 = 150 bar,進樣量= 90 μL,紫外波長= 276 nm;疊層進樣條件:注射次數為12次,疊層時間為0.66 min,色譜峰收集=1(以時間為基礎)。
5.5 普魯卡因
普魯卡因(PC),化學式為4-氨基苯甲酸2-(N,N-二乙胺)乙酯,是一種藥物,用作局部麻醉劑。它阻斷了依賴電壓的鈉離子通道,從而導致對疼痛的敏感性降低。普魯卡因是由4-氨基苯甲酸乙酯(ABE)化學堿催化合成的。這涉及到與2-二乙胺乙醇的酯交換反應。該堿為乙醇酸鈉,可由單質鈉與乙醇反應生成。進行柱篩選以確定純化普魯卡因合成產物的理想選擇性(見圖9)。
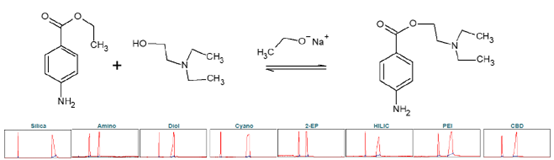
▲ 圖9. 上:普魯卡因合成反應方程,下:Sepmatix 8x SFC篩選結果;從左到右依次為:硅膠、氨基、二醇、氰基、2-EP、HILIC、PEI和CBD;運行時間= 15分鐘。
由于 PC 在固定相中的高保留率,在梯度中增加了一個在 50% 改性劑下 5 分鐘的等度步驟。原則上,每個固定相對普魯卡因的純化都有合適的選擇性(見表5)。普魯卡因在硅膠、PEI和氰基相中具有較高的保留率。由于普魯卡因的基本性質,在篩選條件下,相應峰的峰形較差。二醇相為強前向,不對稱度為0.58;硅膠相為強尾向,不對稱度為 2.73。ABE 后每個固定相的 PC 洗脫。
表5. SFC 條件下不同篩選柱的分辨率值和洗脫順序。
色譜柱 | R | 洗脫順序 |
硅膠 | 16.65 | ABE, PC |
氨基 | 6.54 | ABE, PC |
氰基 | 8.59 | ABE, PC |
二醇基 | 9.16 | ABE, PC |
2-EP | 5.29 | ABE, PC |
HILIC | 5.67 | ABE, PC |
PEI | 3.69 | ABE, PC |
CBD | 7.26 | ABE, PC |
選擇 2-EP 相進行制備純化。硅膠相需要高含量的改性劑,這導致運行時間長,峰形差。2-EP 相顯示樣品的快速洗脫,具有足夠高的分辨率。甲醇改性劑中加入 5% 去離子水和 20mm 氨水作為添加劑。篩選結果表明,改性劑含量為 24 ~ 32% 時, BC 和 PABA 的洗脫效果較好。圖10(上)為改性劑含量為 25% 時,PC 在 2-EP 制備柱上的純化結果。在相同條件下,可采用疊層進樣法對 PC 進行自動化純化(見下圖10)。添加劑的加入顯著改善了 PC 的峰形。
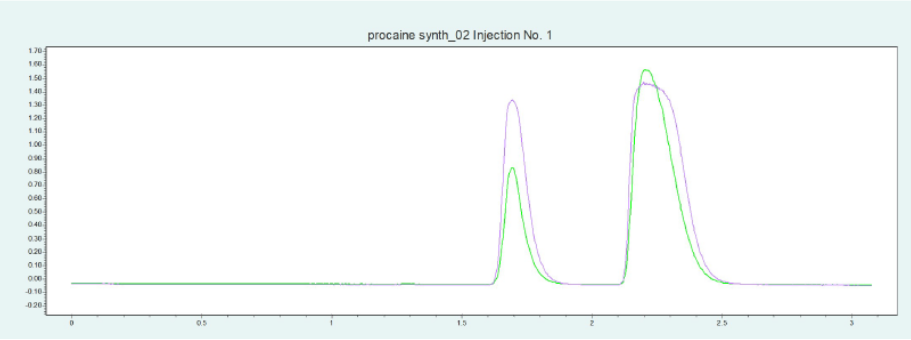
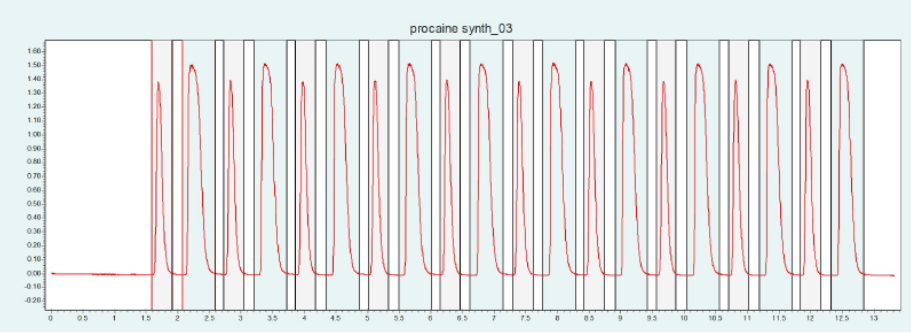
▲ 圖10. 普魯卡因純化的單次進樣(上)和疊層進樣(下);運行條件:流量= 20 mL/min,改性劑=甲醇/水(95/ 5%),含20 mm氨水,改性劑% = 25%,溫度= 40℃,背壓調節器 = 150 bar,進樣量= 100 μL,紫外波長= 220 nm;疊層進樣條件:注射次數= 10次,疊層時間= 1.1 min,色譜峰收集= 2(以時間為基礎)。
6
實驗結論
在進行有機合成后,由于副反應或轉化產物收率不是 100%,雜質通常存在。這些雜質必須除去,特別是在藥品生產中。在本文中,使用 Sepiatec SFC-50 儀器純化了幾種藥物。藥物合成后,使用 Sepmatix 8x SFC 儀器對每種藥物進行色譜柱篩選,以確定純化工藝的最佳色譜柱。最后采用最合適的色譜柱進行制備。合成產物的柱篩選表明,不同的固定相往往具有最佳的純化選擇性。因此,在純化前確定最合適的色譜柱是非常重要的。如實例所示,篩選結果可用于實施一種簡單的制備方法。如果合成混合物不是太復雜,可以使用疊層進樣進行以節約純化時間。




免責聲明
- 凡本網注明“來源:化工儀器網”的所有作品,均為浙江興旺寶明通網絡有限公司-化工儀器網合法擁有版權或有權使用的作品,未經本網授權不得轉載、摘編或利用其它方式使用上述作品。已經本網授權使用作品的,應在授權范圍內使用,并注明“來源:化工儀器網”。違反上述聲明者,本網將追究其相關法律責任。
- 本網轉載并注明自其他來源(非化工儀器網)的作品,目的在于傳遞更多信息,并不代表本網贊同其觀點和對其真實性負責,不承擔此類作品侵權行為的直接責任及連帶責任。其他媒體、網站或個人從本網轉載時,必須保留本網注明的作品第一來源,并自負版權等法律責任。
- 如涉及作品內容、版權等問題,請在作品發表之日起一周內與本網聯系,否則視為放棄相關權利。


 采購中心
采購中心